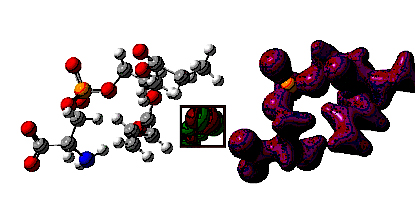|
International Journal of Bioelectromagnetism Vol. 6, No. 2, pp. 33-40, 2004. |
 |
 |
www.ijbem.org |
|
Molecular Dynamics Simulation Kenichi Yamanishia, Lukas Pichla, Yuko Nitahara-Kasaharab, Yoko Aidab aDepartment of computer software, University of Aizu, Aizu-Wakamatsu, Fukushima, Japan Correspondence: L Pichl, University of Aizu, Tsuruga, Ikki, Aizu-Wakamatsu, 965-8580 Japan. Abstract.
Lipid membranes that include various proteins interface the inner and outer space
in cells and nuclei, and play an important role in processes such as cell division
or viral infection. Global electrostatic properties of the system emerge from
interaction of hydrophobic and hydrophilic units in lipids and proteins. Available
experimental techniques cannot directly observe membrane dynamics at the sub-nanometer
level of lipid and peptide components. We therefore study the lipid-lipid and lipid-protein
dynamics by means of computer simulation. The theoretical framework builds on ab initio
calculations of electron probability densities in order to obtain parameters for molecular
dynamics simulation. A coarse grained model approach is adopted, including up to tens of
thousands lipid molecules. Complimentary small-scale atomic-level simulations are performed
for comparison. Dynamics of lipid bilayer including self-assembly process and adhesion
around an a-helix protein fragment are reported. The present work is a step towards
developing a more complex computer simulation framework.
Keywords: Phospholipid molecule; Cell membrane; Ab initio electron density; Langevin equation; Leapfrog algorithm.
1. Introduction Computer simulation of biological membranes is a challenging problem requiring thousands of CPU hours for system of very moderate sizes [Berger et al., 1997; Wohlert and Edholm, 2004]. Recently, various supercomputer centers over the world engaged in this emerging field of science, motivated in part by promising applications to pharmaceutical research [Mashl et al., 2001]. A molecular dynamics (MD) simulation in a reasonably long time interval (~100 ns) using pair-wise interactions is tractable up to the order of 100, 000 particles, which translates at the atomic level into an ensemble of about 1, 000 lipid molecules. Computational benchmarks at such scale suffice in principle for an accurate calibration of lower-resolution aggregate models, which extends the computer simulation in time and space using the coarse-grained model strategy. At present, most of the MD-simulated membranes consist of a single lipid prototype, e.g. the 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) molecule or 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) molecule. As the calibration of simulation techniques has proceeded, more realistic membrane models (for nuclear, plasma, and neural membranes) include not only various phospholipids (choline, ethanolamine, serine, and inositol groups) but also cholesterol, glyco-lipids, and sphingomyelin. The distribution of unsaturated bonds in fatty acid tails of the lipids in the inner part of the membrane, and the curvature effects breaking symmetry of the bilayer should also be accounted for. Lipid molecules in the coarse-grained model are simulated as a functional 3-particle system, which consists of a hydrophilic head and two hydrophobic tails in spatially fixed configuration [Noguchi and Takasu, 2001 and 2002]. A corresponding coarse-grained level of protein description is to represent amino acid side chains by single centers that impose additional forces on peptide backbone [Takada, 1999 and 2001] and interact with the fatty tails of lipids through collective hydrophobic particle density effect. The molecular dynamics framework does not include hydrogen transfer, electronic excitation, ion concentration gradients and other factors. Instead, the effect of some disregarded factors is estimated by introducing a heat bath and stochastic fluctuations in the model. In order to assess the validity of results from numerical experiments, the simulation is repeated in various paths and analyzed collectively. Phenomena studied in computer simulations must be robust against the noise and fluctuations effects. An example of practical interest can be membrane destabilization mechanism as induced by fusion peptides or diffusion of small nonpolar molecules through lipid bilayers. The paper is organized as follows. In Section 2, we discuss the determinants of electron probability density for lipid molecules using density functional theory. Section 3 briefly outlines the structure of coarse-grained model and the MD computational algorithms. In Section 4, we discuss lipid bilayer self-assembly, fluctuations, and interaction with external field induced by peptides. Concluding remarks close the paper in Section 5. 2. Electron density in lipid molecules Electron probability distribution over the lipid molecule is an important determinant of lipid membrane dynamics. The bioelectric potential energy wall of cell or nucleus membrane derives from the wave-functions of lipid and protein components and determines various transport processes at the aggregate scale [Malmuvio and Plonsey, 1995]. For instance, the membrane ionic channels have been well understood and used in body organ simulations [Wei, 1995]. To describe a local phenomenon of lipid membrane destabilization, the electric field of lipid needs to be simulated at the level of functional (head, tail) and atomic components. Electron probability density is low at the hydrophobic lipid tails as compared to the head alcohol group. Figure 1 shows the atomic structure of a phosphatidyl-choline lipid molecule and an isosurface of the electric field computed with the local density functional method using GAUSSIAN 03. The two orthogonal highest occupied and the lowest unoccupied molecular orbitals are located around lipid head as shown in the inset of the figure; electron density on the hydrophobic tails closely follows the bond structure. Table 1 summarizes partial electric charges computed for each atom of this molecule. Figure 1. Phosphatidyl-choline molecule and its electric field isosurface (value 0.1). The inset displays the interleaving structure of the highest occupied and lowest unoccupied molecular orbitals
3. Material and Methods Membrane constituents included in computer simulation are: phosphatidyl-choline, phosphatidyl-ethanolamine, phosphatidyl-serine, phosphatidyl-inositol, sphingo-myelin, phosphoglyceride, and cholesterol. In addition, we include a small accessory viral protein R (Vpr, pdb code 1M8L) with 96 residues. The coordinate files have been downloaded from standard public protein structure databases. The explicit amino-acid side chains are removed; peptide bond lengths are frozen along the backbone. Interaction parameters for the model for each amino acid follow in detail previous work [Takada, 2001; Pichl et al., 2004]. In addition, space distribution of electron charge has been computed with Gaussian 03 using the Becke3lyp density functional method for each membrane component. Membrane self-assembly processes has been simulated with the phosphatidyl-choline ensemble only. 3.1 Coarse grained model The motion of all particles is given by classical mechanics and obeys the set of coupled differential equations of the second order,
where j labels molecules (lipid, protein) and i labels their
components (head, tail, amino-acid). The three coordinates of each
of each vector have been omitted for simplicity. Here mi stands for
the mass of each molecule and ξi is the friction constant. The term
gij(t) denotes Gaussian white noise normalized by system temperature
T, <gij(t)>=0 and
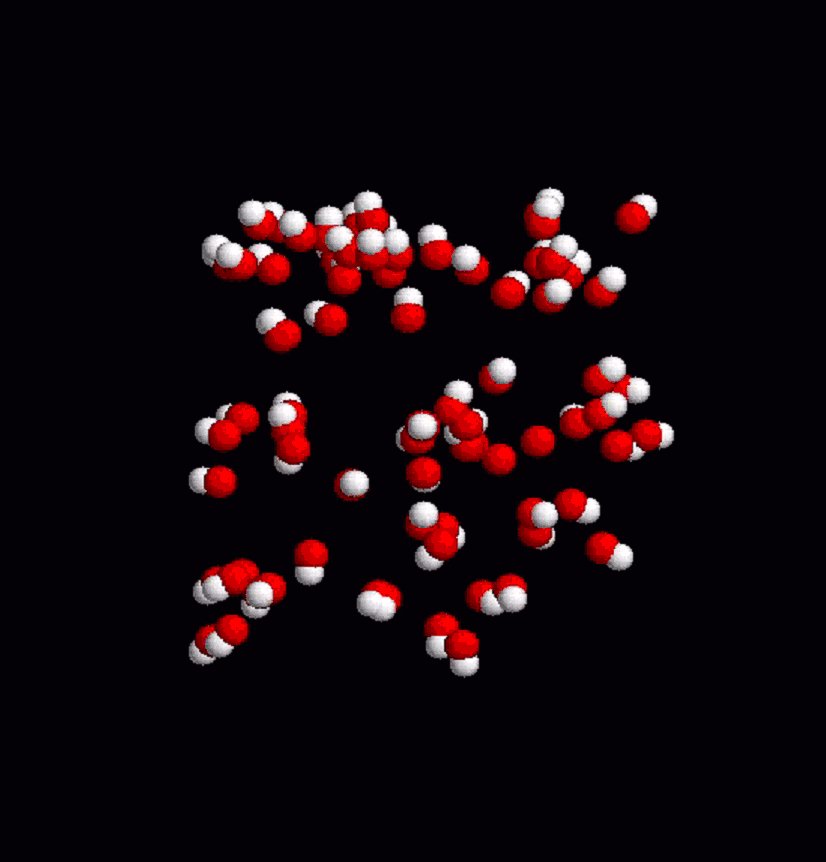
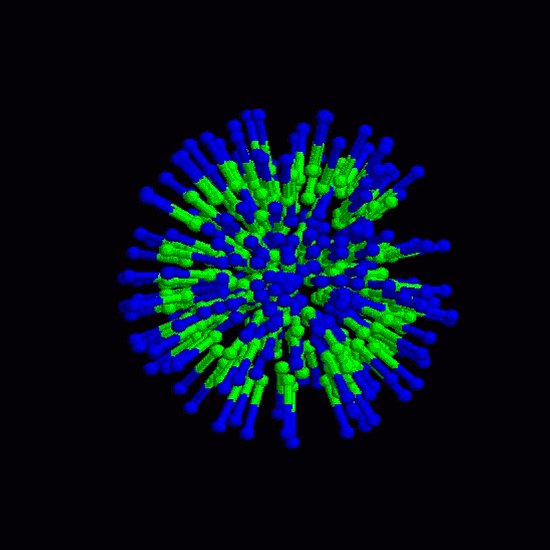
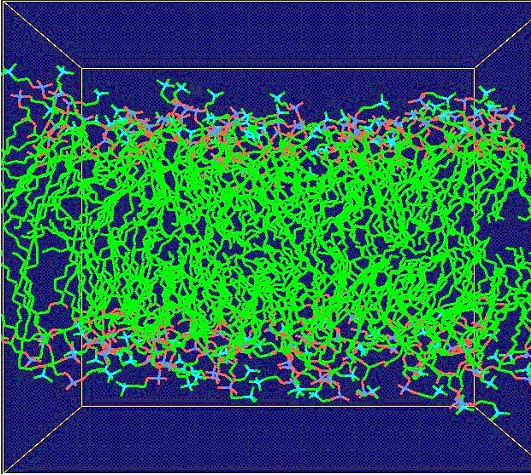
This iterative formula for the phase-space trajectory update is then modified by including the LINCs algorithm [Hess et al., 1997], which ensures that bond lengths are kept fixed as prescribed in the initial configuration. Throughout the coarse-grained model simulations, coordinate files are output in pdb format in regular time interval to be visualized with RASMOL (freeware). The static frames are then encoded into an mpeg video (online version of the journal). Results of molecular dynamics simulation at the atomic level are visualized by using Gromacs trajectory viewer, ngmx, and vide-recorded on the linux platform using VNC (freeware). 4. Results Figure 2 shows a simulation of lipid-Vpr interaction dynamics in the coarse grained model. Lipid molecules are first randomly distributed at high initial velocities. The 3D simulation cell adopts periodic boundary conditions. Subjected to Eq. 1 and the field of Vpr fragment (not displayed in the figure for the sake of clarity), lipid molecules gradually slow down and adhere near the surface of the peptide as a result of the collective interaction potential for hydrophobic particles. Fluctuations in the process result from temperature effects, in which small random forces act on each particle. Figures 3 and 4 show the dynamics of membrane surface and alignment degree. The temperature of the system is set at 25% of the critical value at which the system disintegrates by thermal noise. All animations are downsized for the sake of clarity. Finally, Fig. 5 shows the calibration of coarse-grained model by atomic-level simulation with Gromacs force field for a small homogenous phosphatidyl-choline lipid bilayer. Figure 2. Lipid dynamics in a 3D simulation cell with periodic boundary conditions. Red balls denote hydrophilic heads, white balls represent the two hydrophobic tails. The animation in the online version of the journal shows how the lipid molecules distributed initialy at random slow down in the viscose environment and adapt to a potential field of an a-helix fragment (center of the box, only the lipid particle are shown here). Figure 3. Thermal fluctuations of a membrane surface. The small white spots show hydrophobic lipid tails. Figure 4. Thermal fluctuations of a membrane. The stick bonds represent the internal degree of alignment in the lipid belayer. 5. Conclusions We have built a computational hybrid model that includes collective density potentials for hydrophobic behavior and solvent effects, and studied lipid-protein dynamics in the coarse-grained model framework. Electric field of relevant membrane lipids has been computed by applying the density functional method and used to determine the interaction parameters of the coarse grained model. Our molecular dynamics calculation is based on the leap-frog algorithm adapted to the LINCS fixed constraint solver. Within this framework, we have observed lipid adhesion along an a-helix fragment and fluctuations of membrane surface. Atomic-level simulation has been performed for calibration and a good level of agreement is found between the two approaches. Figure 5. Molecular dynamics of homogenou lipid bilayer at atomic resolution level with the Gromacs force fields. Phosphatidyl-choline molecules are displayed inside the simulation box Acknowledgements Lukas Pichl acknowledges partial support of this work by a Grant-in-Aid from the Japan Society for the Promotion of Science. References
Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophysical Journal 72: 2002-2013, 1997. Mashl R J, Scot H L, Subramaniam S, Jakobsson E. Molecular Simulation of Dioleoylphosphatidylcholine Lipid Bilayers at Differing Levels of Hydration. Biophysical Journal 81(6): 3005-3015, 2001. Hess B, Bekker H, Berendsen H, Fraaije J. LINCS: A linear constraint solver for molecular simulations, Journal of Computational Chemistry 18: 1463-1472 (1997). Malmivuo J, Plonsey R. Bioelectromagnetism: Principles and Application of Bioelectric and Biomagnetic Fields. Oxford University Press, New York, 1995. Noguchi H, Takasu M. Self-assembly of amphiphiles into vesicles, Physical Review E 64: 041913: 1-7, 2001. NoguchiH, Takasu M. Adhesion of Nanoparticles to Vesicles, Biophysical Journal 93: 299-308, 2002. Takada S, Luthey-Schulten Z, Walynes P G. Folding dynamics with nonadditive forces, Journal of Chemical Physics 110: 11616-29 (1999). Takada S, Protein folding simulation with solvent-induced force field, PROTEINS: Structure, Function and Genetics 42: 85-98 (2001). Pichl L, Yamanishi K, Nitahara Y, Aida Y. Three dimensional space interaction model for HIV-1 accessory protein Vpr and membrane lipid molecules, Journal of Three Dimensional Images, 2004, in print. Wei D, Okazaki O, Harumi K, Harasawa E, Hosaka H.Comparative simulation of Excitation and body surface electrocardiogram with isotropic and anisotropic computer heart models, IEEE Transactions on Biomedical Engineering 42:343-357, 1995. Wohlert J, Edholm O. The Range and Shielding of Dipole-Dipole interactions in Phospholipid Bilayers. Biophysical Journal 87:2433-2445, 2004.
|